ORILISSA 150 mg QD
Demonstrated safety profile
Adverse reactions in ≥5% of subjects and their corresponding discontinuation rates1-5*

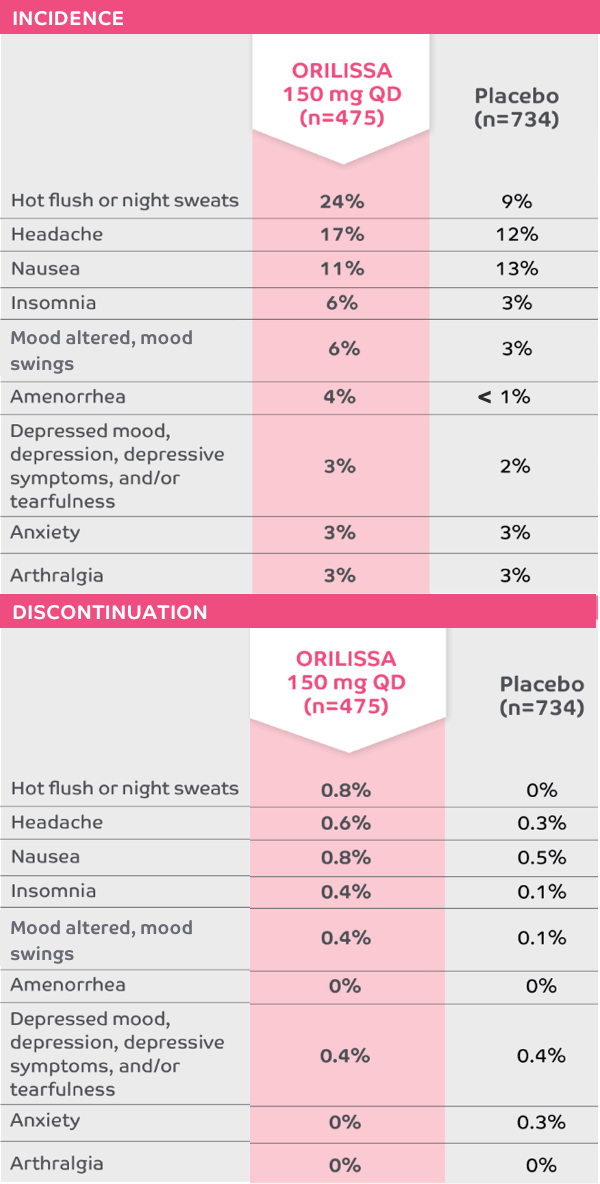
The most common serious adverse events reported with ORILISSA in ELARIS EM-1 and EM-2 included appendicitis (0.3%), abdominal pain (0.2%), and back pain (0.2%).1
0.2% of patients treated with ORILISSA 150 mg QD discontinued therapy due to serious adverse reactions compared to 0.5% receiving placebo.1
*Adverse reactions that occurred at a greater incidence than placebo in either ORILISSA dose group in ELARIS EM-1 and ELARIS EM-2.
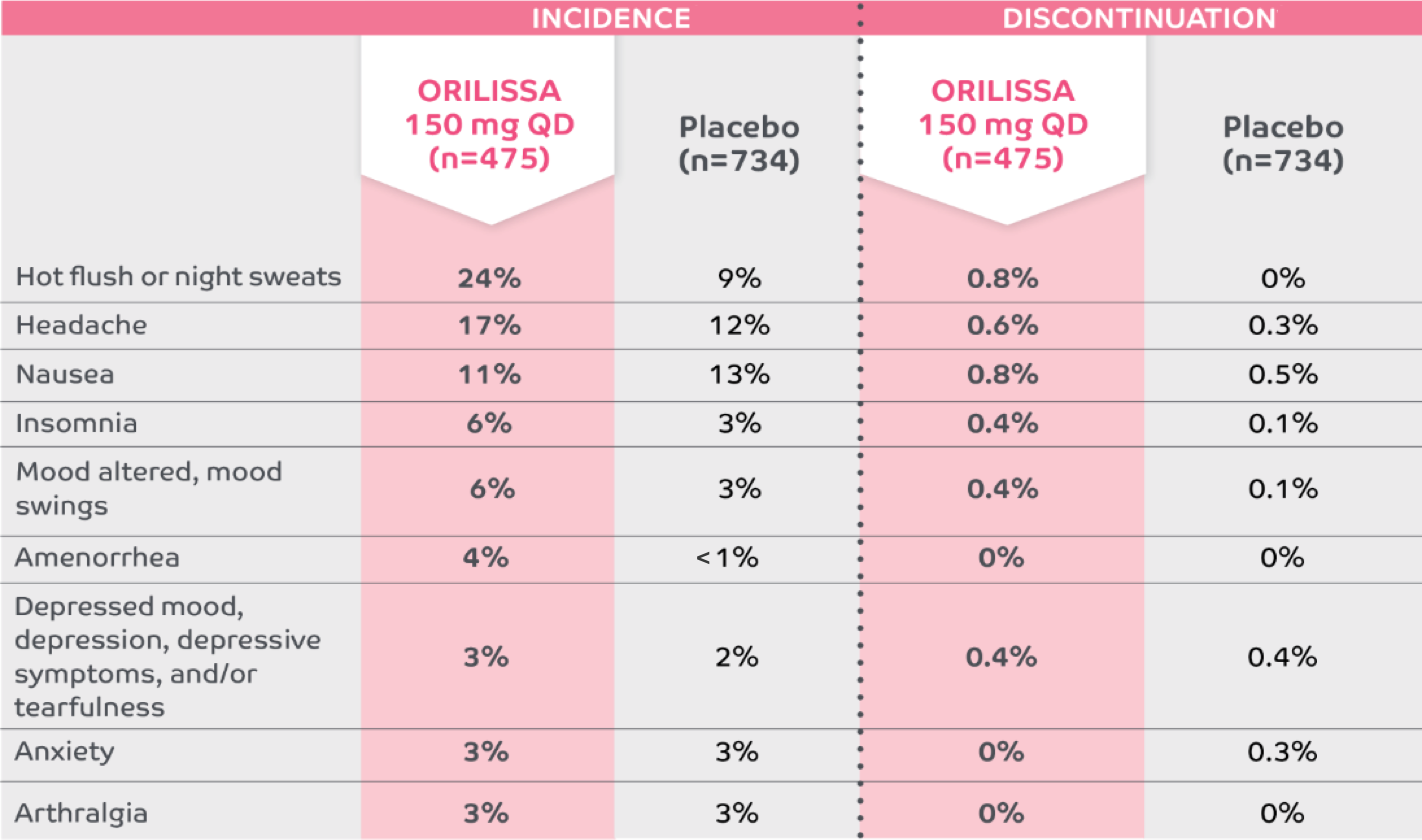
Low discontinuation rates due to any adverse reaction1,5
ORILISSA 150 mg QD
Placebo
The most common serious adverse events reported with ORILISSA in ELARIS EM-1 and EM-2 included appendicitis (0.3%), abdominal pain (0.2%), and back pain (0.2%).1
0.2% of patients treated with ORILISSA 150 mg QD discontinued therapy due to serious adverse reactions compared to 0.5% receiving placebo.1
*Adverse reactions that occurred at a greater incidence than placebo in either ORILISSA dose group in ELARIS EM-1 and ELARIS EM-2.

ORILISSA 200 mg BID
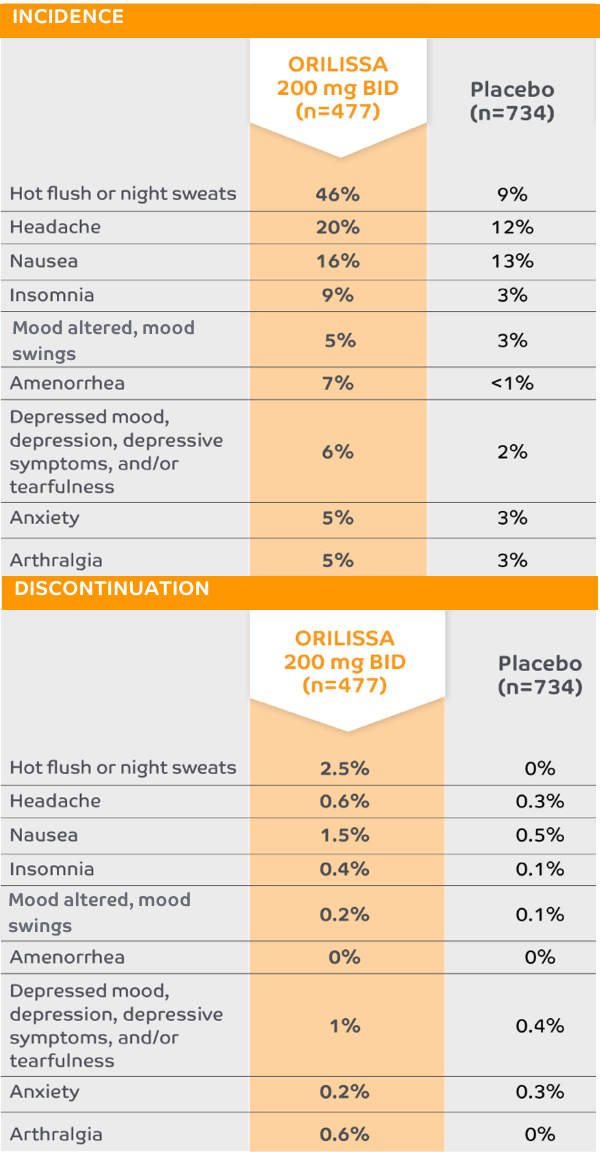
The most common serious adverse events reported with ORILISSA in ELARIS EM-1 and EM-2 included appendicitis (0.3%), abdominal pain (0.2%), and back pain (0.2%).1
0.2% of patients treated with ORILISSA 200 mg BID discontinued therapy due to serious adverse reactions compared to 0.5% received placebeo.1
*Adverse reactions that occurred at a greater incidence than placebo in either ORILISSA dose group in ELARIS EM-1 and ELARIS EM-2.
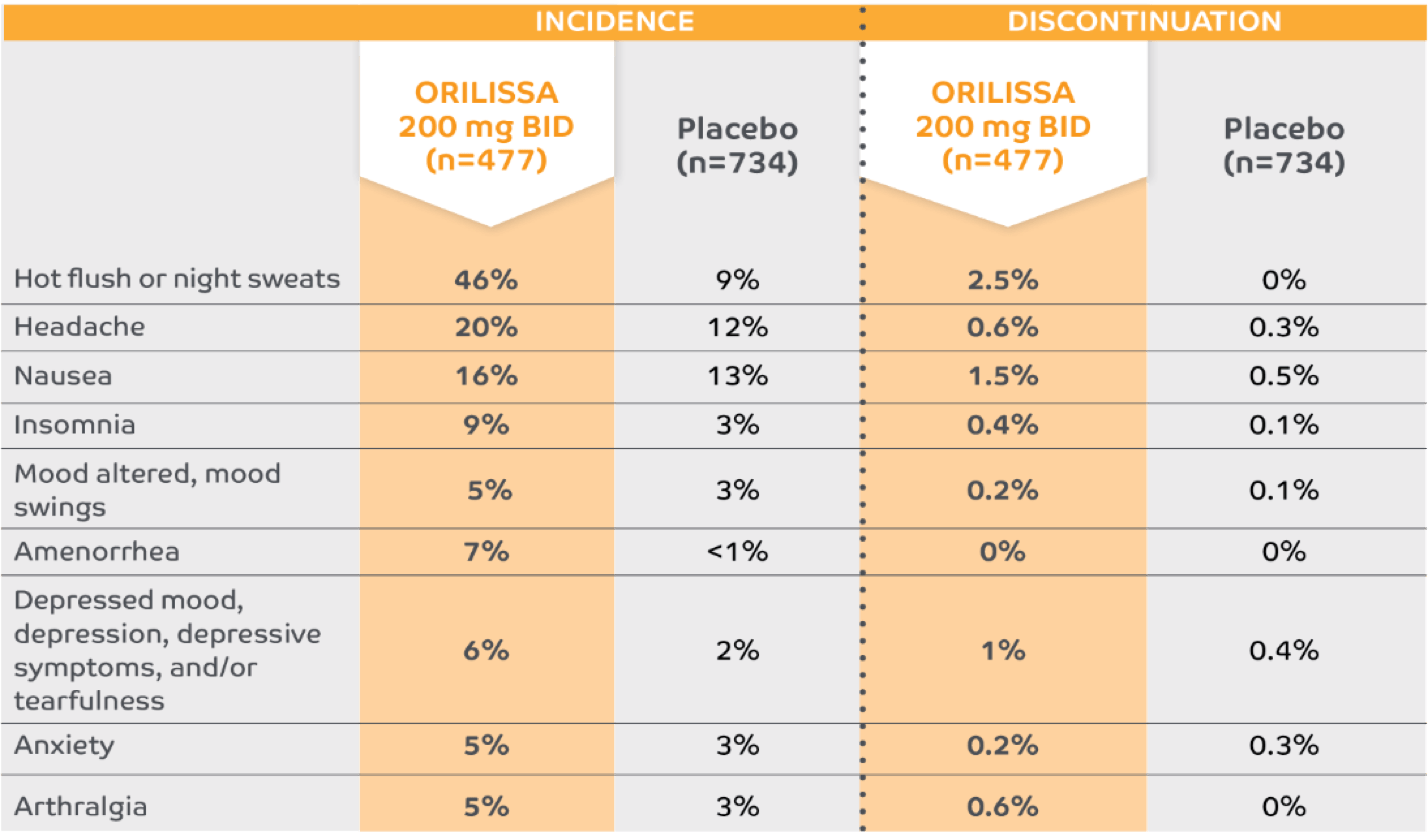
The most common serious adverse events reported with ORILISSA in ELARIS EM-1 and EM-2 included appendicitis (0.3%), abdominal pain (0.2%), and back pain (0.2%).1
0.2% of patients treated with ORILISSA 200 mg BID discontinued therapy due to serious adverse reactions compared to 0.5% receiving placebo.1
*Adverse reactions that occurred at a greater incidence than placebo in either ORILISSA dose group in ELARIS EM-1 and ELARIS EM-2.
Low discontinuation rates due to any adverse reaction1,5
ORILISSA 200 mg BID
Placebo
The most common serious adverse events reported with ORILISSA in ELARIS EM-1 and EM-2 included appendicitis (0.3%), abdominal pain (0.2%), and back pain (0.2%).1
0.2% of patients treated with ORILISSA 200 mg BID discontinued therapy due to serious adverse reactions compared to 0.5% receiving placebo.1
*Adverse reactions that occurred at a greater incidence than placebo in either ORILISSA dose group in ELARIS EM-1 and ELARIS EM-2.
The most common serious adverse events reported with ORILISSA in ELARIS EM-1 and EM-2 included appendicitis (0.3%), abdominal pain (0.2%), and back pain (0.2%).1
0.2% of patients treated with ORILISSA 200 mg BID discontinued therapy due to serious adverse reactions compared to 0.5% receiving placebo.1
*Adverse reactions that occurred at a greater incidence than placebo in either ORILISSA dose group in ELARIS EM-1 and ELARIS EM-2.
Low discontinuation rates due to any adverse reaction1,5
ORILISSA 200 mg BID
Placebo
Contraindications1
ORILISSA is contraindicated in women:
- Who are pregnant. Exposure to ORILISSA early in pregnancy may increase the risk of early pregnancy loss
- With known osteoporosis because of the risk of further bone loss
- With severe hepatic impairment
- Taking inhibitors of organic anion transporting polypeptide (OATP) 1B1 that are known or expected to significantly increase elagolix plasma concentrations
- With known hypersensitivity reaction to ORILISSA or any of its inactive components. Reactions have included anaphylaxis and angioedema
Indication and Important Safety Information
INDICATION1
ORILISSA® (elagolix) is indicated for the management of moderate to severe pain associated with endometriosis.
IMPORTANT SAFETY INFORMATION1
CONTRAINDICATIONS
- ORILISSA is contraindicated in women who are pregnant (exposure to ORILISSA early in pregnancy may increase the risk of early pregnancy loss), in women with known osteoporosis or severe hepatic impairment, or with concomitant use of strong organic anion transporting polypeptide (OATP) 1B1 inhibitors (e.g., cyclosporine and gemfibrozil).
WARNINGS AND PRECAUTIONS
Bone Loss
- ORILISSA causes a dose-dependent decrease in bone mineral density (BMD), which is greater with increasing duration of use and may not be completely reversible after stopping treatment.
- The impact of ORILISSA-associated decreases in BMD on long-term bone health and future fracture risk is unknown. Consider assessment of BMD in patients with a history of low-trauma fracture or other risk factors for osteoporosis or bone loss, and do not use in women with known osteoporosis.
- Limit the duration of use to reduce the extent of bone loss.
Change in Menstrual Bleeding Pattern and Reduced Ability to Recognize Pregnancy
- Women who take ORILISSA may experience a reduction in the amount, intensity, or duration of menstrual bleeding, which may reduce the ability to recognize the occurrence of pregnancy in a timely manner. Perform pregnancy testing if pregnancy is suspected, and discontinue ORILISSA if pregnancy is confirmed.
Suicidal Ideation, Suicidal Behavior, and Exacerbation of Mood Disorders
- Suicidal ideation and behavior, including one completed suicide, occurred in subjects treated with ORILISSA in the endometriosis clinical trials.
- ORILISSA users had a higher incidence of depression and mood changes compared to placebo and ORILISSA users with a history of suicidality or depression had an increased incidence of depression. Promptly evaluate patients with depressive symptoms to determine whether the risks of continued therapy outweigh the benefits. Patients with new or worsening depression, anxiety, or other mood changes should be referred to a mental health professional, as appropriate.
- Advise patients to seek immediate medical attention for suicidal ideation and behavior. Reevaluate the benefits and risks of continuing ORILISSA if such events occur.
Hepatic Transaminase Elevations
- In clinical trials, dose-dependent elevations of serum alanine aminotransferase (ALT) at least 3 times the upper limit of the reference range occurred with ORILISSA.
- Use the lowest effective dose and instruct patients to promptly seek medical attention in case of symptoms or signs that may reflect liver injury, such as jaundice.
- Promptly evaluate patients with elevations in liver tests to determine whether the benefits of continued therapy outweigh the risks.
Reduced Efficacy with Estrogen-Containing Contraceptives
- Based on the mechanism of action of ORILISSA, estrogen-containing contraceptives are expected to reduce the efficacy of ORILISSA. The effect of progestin-only contraceptives on the efficacy of ORILISSA is unknown.
- Advise women to use non-hormonal contraceptives during treatment and for one week after discontinuing ORILISSA.
ADVERSE REACTIONS
- The most common adverse reactions (>5%) in clinical trials included hot flushes and night sweats, headache, nausea, insomnia, amenorrhea, anxiety, arthralgia, depression-related adverse reactions, and mood changes.
These are not all the possible side effects of ORILISSA.
Safety and effectiveness of ORILISSA in patients less than 18 years of age have not been established.
US-ORIL-200330
For more information, please click here for full Prescribing Information.
References:
1. ORILISSA [package insert]. North Chicago, IL: AbbVie Inc. 2. Data on file. ABVRRTI68989. 3. Taylor HS, Giudice LC, Lessey BA, et al. Treatment of endometriosis-associated pain with elagolix, an oral GnRH antagonist. N Engl J Med. 2017;377(1):28-40. 4. Data on file. ABVRRTI68861. 5. Goolsby MA, Boniquit N. Bone health in athletes: the role of exercise, nutrition, and hormones. Sports Health. 2017;9(2):108-117. 6. Langdahl B, Ferrari S, Dempster DW. Bone modeling and remodeling: potential as therapeutic targets for the treatment of osteoporosis. Ther Adv Musculoskel Dis. 2016;8(6):225-235. 7. Karlsson C, Obrant KJ, Karlsson M. Pregnancy and lactation confer reversible bone loss in humans. Osteoporos Int. 2001;12(10):828-834. 8. Lupron Depot [package insert]. North Chicago, IL: AbbVie Inc; 2018.
9. Clark MK, Sowers MF, Levy B, Nichols S. Bone mineral density loss and recovery during 48 months in first-time users of depot medroxyprogesterone acetate. Fertil Steril. 2006;86(5):1466-1474. 10. Data on file. ABVRRTI68937. 11. Data on file. ABVRRTI68942.



